




Thesis by Thomas Pigeon: « Méthodes d’échantillonnage d’évènements rares et machine learning pour l’étude des mécanismes de réaction catalytiques » (Rare event sampling methods and machine learning for the study of catalytic reaction mechanisms).
Understanding the chemical properties of the supports (alumina-gamma in particular) and active phases of heterogeneous catalysts is a challenge that requires a detailed atomic scale description of systems and the quantification of events that are rare on this scale: chemical reactions.
Quantum simulation appears to be a suitable tool for trying to overcome this challenge. However, continuous improvement of numerical methodologies and atomistic models is required to determine the complex structure of active sites, on the one hand, and their reactivity (rate constant), on the other. By covering all these aspects, this PhD research has provided answers to this dual challenge, while exploring the contributions of machine learning (ML) to the understanding of the active sites.
Transition State Theory (TST) inevitably overestimates reactivity. The latter is very often coupled with a harmonic approximation of the potential energy surface (hTST), implying a second source of uncertainty related to the rate constant calculation. To overcome these difficulties, an alternative approach was implemented to calculate reaction rate constants directly. This approach combines a rare-event simulation method AMS1 with AIMD2, implemented with quantum software (such as VASP3) widely used by the community (Figure 1a).
First of all, ML tools (SVM [1], Autoencoders [2]) made it possible to identify the collective variables (CVs) used to automatically define the progress of AMS during a reaction (Figure 1b). They were also used to build an interatomic force field that accelerated AIMD calculations. Using SVM, AMS and AIMD, the rate constants of rotation and dissociation of a water molecule on the surface (100) of alumina-gamma[1] were evaluated. These constants turn out to be up to two orders of magnitude lower than those obtained with the hTST approach, revealing the errors generated by the latter [1].
In parallel, with a view to a more detailed description of the active sites present on alumina-gamma crystallites, several complex facet [3] and edge [4] models were developed. These were used to provide a more detailed interpretation of the proton’s experimental NMR4 spectra (Figure 1c). Ab initio calculations led to the identification of previously undocumented sites located on edges.
This research, conducted within the dual context of the INRIA-IFPEN framework agreement and the ENSL-IFPEN ROAD4CAT chair, is now being continued as part of the MAMABIO project, attached to PEPR B-BEST.
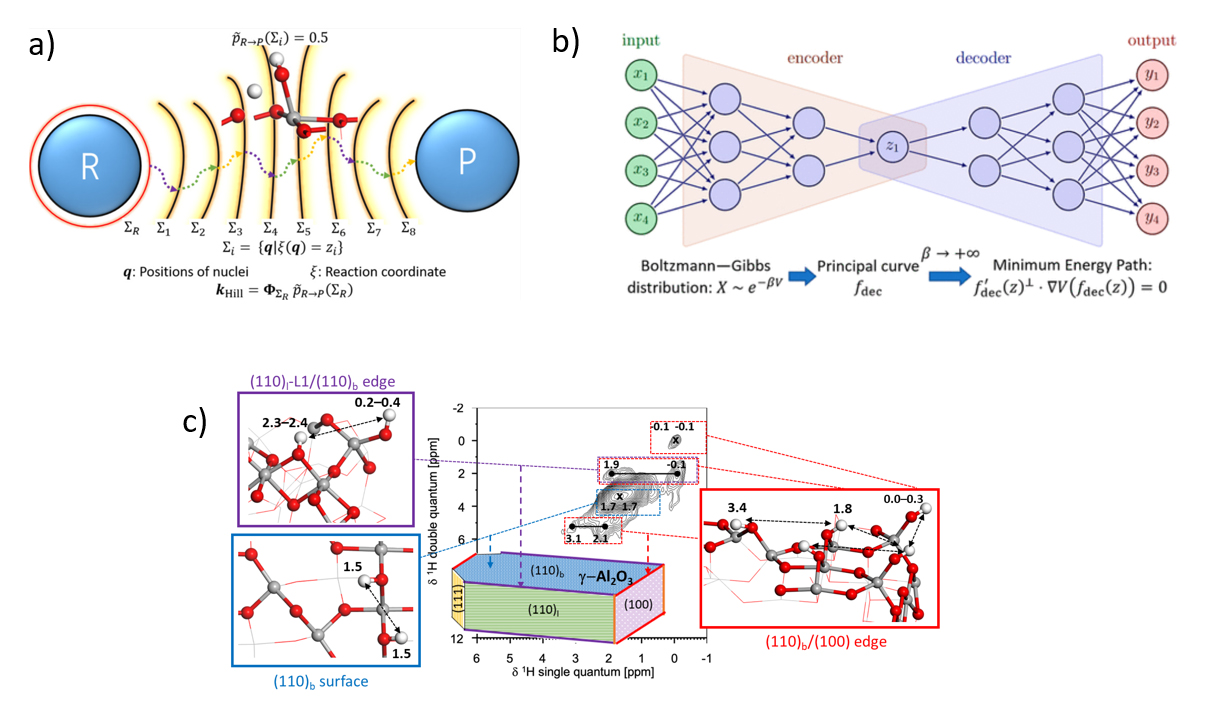
a) Diagram of the estimator constructed by AMS to estimate rate constants,
b) Illustration of an auto-encoder and the physical meaning of the CVs learned by this method,
c) Assignment, based on atomistic models, of 2D 1H NMR signals to hydroxyls of surface and edge sites of gamma alumina.
1- Adaptive Multilevel Splitting
2- Ab Initio Molecular Dynamics
3- Vienna Ab-initio Simulation Package, https://www.vasp.at/
4- Nuclear Magnetic Resonance
References:
-
T. Pigeon, G. Stoltz, M. Corral-Valero, A. Anciaux-Sedrakian, M. Moreaud, T. Lelièvre, P. Raybaud, Computing Surface Reaction Rates by Adaptive Multilevel Splitting Combined with Machine Learning and Ab Initio Molecular Dynamics, J. Chem. Theory Comput. 2023, 19, 12, 3538–3550
>> https://doi.org/10.1021/acs.jctc.3c00280
-
T. Lelièvre, T. Pigeon, G. Stoltz, W. Zhang, Analyzing Multimodal Probability Measures with Autoencoders, J. Phys. Chem. B, 2024, 128, 11, 2607–2631
>> https://doi.org/10.1021/acs.jpcb.3c07075
-
T. Pigeon, C. Chizallet, P. Raybaud, Revisiting γ-alumina surface models through the topotactic transformation of boehmite surfaces, J. Catal. 2022, 405, 140-151
>> https://doi.org/10.1016/j.jcat.2021.11.011
-
A. T. F. Batista, T. Pigeon, J. Meyet, D. Wisser, M. Rivallan, D. Gajan, L. Catita, F. Diehl, A-S. Gay, C. Chizallet, A. Lesage, P. Raybaud, Structure, Location, and Spatial Proximities of Hydroxyls on γ-Alumina Crystallites by High-Resolution Solid-State NMR and DFT Modeling: Why Edges Hold the Key, ACS Catal. 2023, 13, 10, 6536–6548
>> https://doi.org/10.1021/acscatal.3c00495
Scientific contacts: thomas.pigeon@ifpen.fr and Pascal Raybaud